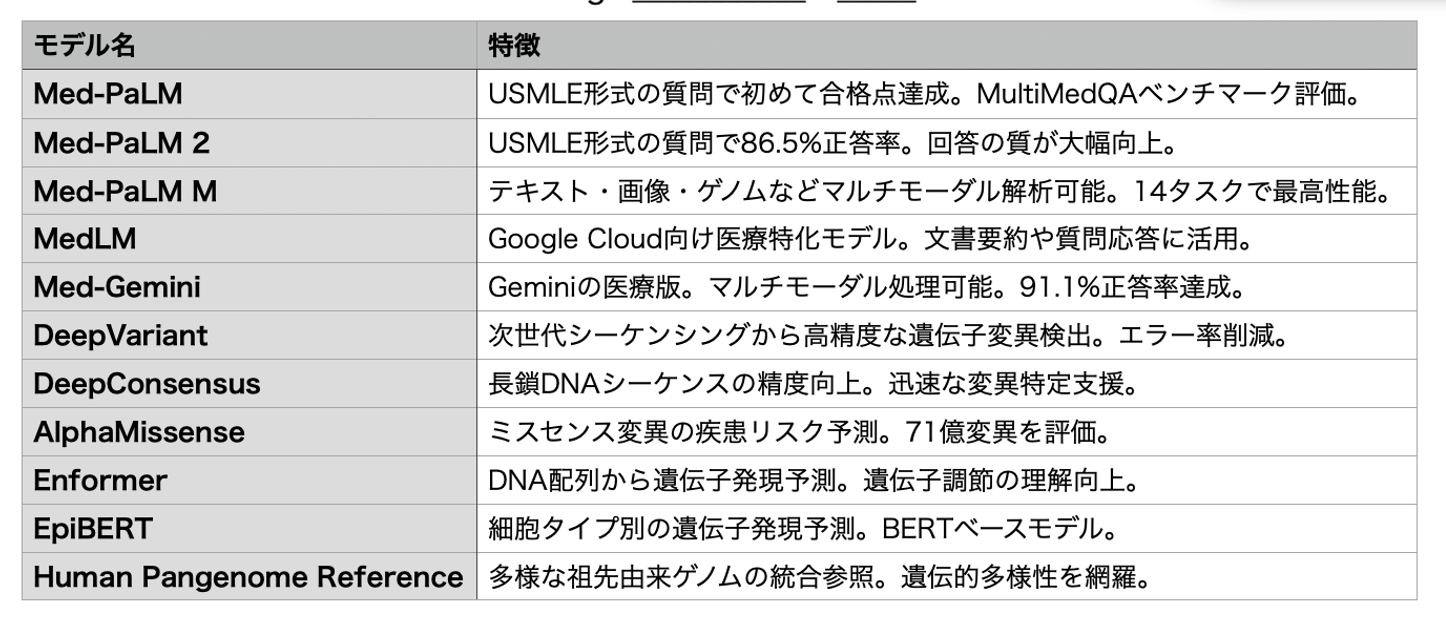
Deepmindを中心にGoogleの様々な研究所から発表されてきた生命科学に関する生成AIモデルの代表は2024年のノーベル賞を受賞したAlphaFoldだが、このブログでもいくつか紹介したように、実際には実に多くのモデルが発表されており、眺めてみるとゲノムを含むあらゆる情報を統合する明確な方向性が見え、医学医療が大きく変革させる流れを作ろうとしているのがわかる(主なものを表にまとめてみた)。
AIの独占禁止が重要な課題になっているが、これらが全てGoogleにつながっているのかと思うと、どうしていいのかわからなくなるほどだ。
そしてこの表に、ゲノムの意味を知るためのもう一つ大きなモデル、AlphaGenome が Google DeepMind から1月28日 Nature に発表された。タイトルは「Advancing regulatory variant effect prediction with AlphaGenome(遺伝子調節変異の効果を推測のための先進モデルAlphaGenome)」だ。
私たち世代は、ゲノム解読が進み、配列ベースで遺伝子発現調節を調べる様々なテクノロジーが進むのを驚きとともに体験した。おそらく今の研究者がノーザンブロッティングを使うことは組織学以外にはないだろう。RNAseq、ChipSeq、Atac-seq など考えてみると全て機能をゲノムへと集約させるために開発されている。AlphaGenomeではヒトやマウスで蓄積されたこれらのデータを学習させて、ゲノムの違いから生成される機能変化を予測する統合モデルを目指している。
基本的には表にも挙げた GoogleDeepmind によって開発された Enformer モデルと目的は同じだが、最も大きな違いは Enformer が100Kb前後のゲノムを入力していたのに対して、AlphaGenome は1Mbという大きな領域を入力して1塩基単位で比較できるようにしている点だ。この1Mbと言う大きな領域を一つのセンテンスとして扱うのは Arc研究所と NVIDIA が開発している Evo と同じで、畳み込みにより一塩基レベルで1Mbという大きな領域を比較出来るようにして、この1Mbについて、これまでの研究が蓄積してきたRNA-seq、Atac-seq、CAGE、 Chip-seq、Splice-site、更にはHiCのようなゲノム領域内の相互作用まで学習させている。
当然このモデルで予測されるのはゲノムの配列がどの細胞でどのように働いているかで、異なる細胞の発現データとして得られた5930ヒトゲノム配列と、1128マウスゲノム配列として学習されている。わかりやすく言うと、大きなDNA配列をB細胞で見たらどうか、グリア細胞で見たらどうかという見方が学習され、予測できるようにしている。従って、各細胞レベルで見ると、Atac-seq として読まれた部分だけでなく、読まれなかったクロマチンが閉じている部分は、文章の中でマスクされた単語と同じで、マスクされているということ自体が情報になっている。従って、各細胞種は系統樹のような形でアウトプットされてくる。
例えばFig6では、T細胞白血病のTAL1発現領域の変異を予測させる課題を行っているが、発現に関わる様々なモダリティーが合わさった細胞即ちT細胞白血病から、それぞれのモダリティーのアウトプットが異なる様々な細胞が系統樹のように示されてくる。その結果、特定のゲノム変異が白血病に関わること、また肝臓ガン(HepG2細胞)ではこの配列の読まれ方が全く異なることもわかる。
畳み込みとTransformerをアーキテクチャーとして持つ他のモデル、Enformer や Borzoi だけでなく、発現量、エンハンサー活性、クロマチン構造と言ったそれぞれの機能を調べるモデルと予測機能を比較しているが、ほぼ完勝(差は大きくないが)で、一つの配列から異なる細胞種での多くの情報をかなり予測できるようになっていることがわかる。また Zero shot で全く新しい配列を与えてスプライシングを予測させるタスクでも、かなり使えることがわかる。
このような高い性能が AlphaGenome で可能になった背景を調べるため、機能を落として同じ予想課題を行わせる実験を行い、1Mbのように長いDNA配列を学習させること、そして一度に様々なモダリティーを学習させることで予測効率が高まることも示している。
以上が結果で、今後自分が対応している患者さんのゲノムからどこまで診断上の重要な情報が得られるのか、それぞれの研究者が例えば病気リスクのGWAS解析の結果を AlphaGenome で調べることで、AlphaFold のように重要なツールになっていくのだと思う。
もう現役ではない私がこのような作業に関わることはない。それでもこの論文を読んで大きな興奮に襲われたのは、2024年にこのブログで紹介した同じく畳み込みとTransformerをアーキテクチャーとして使っているEvoモデル(https://aasj.jp/news/watch/25610)を念頭に読んでみたからだ。どちらも1MbのDNA入力を畳み込みで比較できるモデルだが、Evo1、Evo2は地球上の種の持つDNAをそのまま学習して、新しい配列をデザインすることを目的としている。即ち進化そのもので発生したコンテクストを学び新しいコンテクストを生成することを目的にしている。一方AlphaGenomeは同じDNA入力について、ゲノム解読以来の研究データを学習させ、新しい研究データを生成することを目指している。即ち人智によるゲノム理解を学習させている。とすると、この二つが統合される方向に行くのは自然の成り行きで、例えばEvo2にデザインされたイントロン配列をインプットして、新しい人工配列を生成させ、その意味をAlphaGenomeで調べることができるようになる。逆にAlphaGenomeで理解できている配列をEvo2にインプットしてよく似た機能を保つ新しい配列を生成できる。生物学が神の領域に近づくとよく言われるのはデザインが可能になったときだが、進化というデザインをコンピュータ上で新たに生成することが出来る時代がきたと思うと、これを目の当たりに出来た興奮は冷めない。


1.Evo2にデザインされたイントロン配列をインプットして、新しい人工配列を生成させ、その意味をAlphaGenomeで調べる。
2.AlphaGenomeで理解できている配列をEvo2にインプットしてよく似た機能を保つ新しい配列を生成する
imp.
進化というデザインをコンピュータ上で新たに生成することが出来る時代がきた!